

Identification of N-Terminally Diversified GLP-1R Agonists Using Saturation Mutagenesis and Chemical Design
Identification of N-Terminally Diversified GLP-1R Agonists Using Saturation Mutagenesis and Chemical Design
ACS Chem. Biol. 2021, 16, 1, 58–66
Publication Date:December 14, 2020
https://doi.org/10.1021/acschembio.0c00722
Chelsea K. Longwell, Stephanie Hanna, Nina Hartrampf, R. Andres Parra Sperberg, Po-Ssu Huang, Bradley L. Pentelute, and Jennifer R. Cochran
Abstract
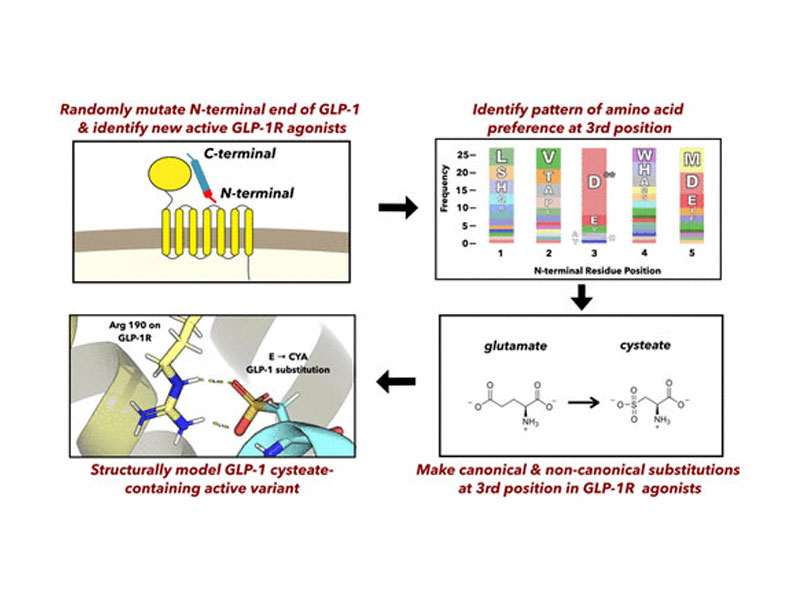
The glucagon-like peptide 1 receptor (GLP-1R) is a class B G-protein coupled receptor (GPCR) and diabetes drug target expressed mainly in pancreatic β-cells that, when activated by its agonist glucagon-like peptide 1 (GLP-1) after a meal, stimulates insulin secretion and β-cell survival and proliferation. The N-terminal region of GLP-1 interacts with membrane-proximal residues of GLP-1R, stabilizing its active conformation to trigger intracellular signaling. The best-studied agonist peptides, GLP-1 and exendin-4, share sequence homology at their N-terminal region; however, modifications that can be tolerated here are not fully understood. In this work, a functional screen of GLP-1 variants with randomized N-terminal domains reveals new GLP-1R agonists and uncovers a pattern whereby a negative charge is preferred at the third position in various sequence contexts. We further tested this sequence–structure–activity principle by synthesizing peptide analogues where this position was mutated to both canonical and noncanonical amino acids. We discovered a highly active GLP-1 analogue in which the native glutamate residue three positions from the N-terminus was replaced with the sulfo-containing amino acid cysteic acid (GLP-1-CYA). The receptor binding and downstream signaling properties elicited by GLP-1-CYA were similar to the wild type GLP-1 peptide. Computational modeling identified a likely mode of interaction of the negatively charged side chain in GLP-1-CYA with an arginine on GLP-1R. This work highlights a strategy of combinatorial peptide screening coupled with chemical exploration that could be used to generate novel agonists for other receptors with peptide ligands.